In 2011, the U.S. Food and Drug Administration (FDA) issued a Guidance for Industry – Process Validation: General Principles and Practices. It highlighted a third validation stage goal of continued process verification (CPV) for:
…continual assurance that the process remains in a state of control (the validated state) during commercial manufacture. A system or systems for detecting unplanned departures from the process as designed is essential to accomplish this goal.

Zuwei Jin
Senior Life Sciences Consultant
The impacts are around three main areas: process development, engineering design practice, and manufacturing operation.
He explained that the FDA started to request more and more statistical analysis for Pharmaceutical Quality/Chemistry Manufacturing and Controls (CMC) submission and science-based proof for proving that the process is in a state of control.
Zuwei has had conversations with product development (PD) scientists, statisticians, and manufacturing operations personnel from several of the world’s leading pharmaceutical companies. To address these requests for increased statistical analysis and proof, initiatives such as quality by design (QbD), design of experiments (DOE) and CPV are clearly where manufacturers in the Life Sciences industry are headed with their process development and manufacturing operations.
For process development labs, popular initiatives include QbD, CPV, DOE, Risk assessment (RA), critical process parameters (CPP), critical quality attributes (CQA) and critical performance attributes (CPA). The overall project and engineering effort benefits from the risk assessment information from stage 1 of the process validation guidelines—that is, adopting a QbD approach.
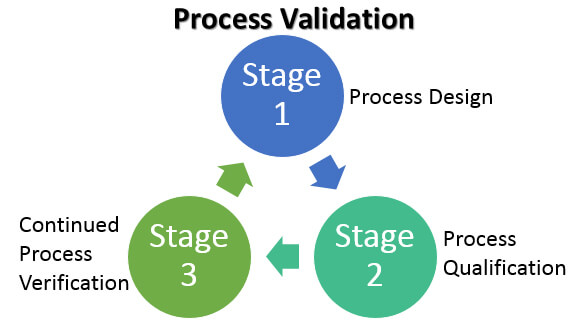
Process Validation Lifecycle
Zuwei shared that more pharmaceutical companies and engineering firms have started to take a risk-based approach such as ASTM E2500 (Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment) for their process validation-stage 2, equipment and facility qualification.
Click to enlarge
Continued process verification (CPV) now needs to be implemented in manufacturing operation (stage 3 in the validation lifecycle). Manufacturing intelligence such what is found in DeltaV Batch Analytics are emerging to meet the data analysis demands now required for stage 3 and may soon become a required component for process control platforms for regulated industries such as the Life Science industry.
Zuwei closed his thoughts with me noting that managing the data and documentation through the validation lifecycle is key in effectively applying the guideline in practice. Early adoption of a level 3 manufacturing execution system (MES) including a document control and archive (DCA) platform will greatly benefit the practice and significantly improve the timelines of the validation and drug development lifecycles.
You can connect and interact with other Life Sciences and manufacturing execution system experts in the Life Sciences and Operations Management groups in the Emerson Exchange 365 community.
